
The Development Alignment Gap

Why Clinical Development Programs Fail Before FDA Ever Reviews the Data
A Strategic Perspective on Evidence Generation, Regulatory Alignment, and Development Success
Denis Katz, MD
Founder, Salience Clinical LLC
Executive Summary
Over the course of my career, I've participated in and advised development programs across pharmaceuticals, biologics, regenerative medicine, diagnostics, neuroscience and many other therapeutic areas. One pattern keeps showing up regardless of the therapeutic area.
The programs that struggled were rarely the ones with the weakest science. More often, they generated evidence that didn't quite match what regulators, clinicians, and payers needed to make a decision. The programs that succeeded had usually done something harder: they'd stayed aligned with those stakeholders the whole way through. Approval, in my experience, isn't the result of one breakthrough decision. It's the accumulation of thousands of smaller ones and every single one of them can quietly open a gap between what a sponsor is building and what a decision-maker needs to see.
I call this the Development Alignment Gap: the disconnect between the evidence sponsors intend to generate and the evidence decision-makers require. It's one of the most important, and most underappreciated, drivers of development risk. It shows up as protocol amendments. Regulatory delays. Trials that fail not because the drug didn't work, but because the trial couldn't prove it did. Costs that balloon past what the science alone would justify.
The numbers back this up. Across the most recent decade of phase-transition data, only around 28–29% of programs that enter Phase II make it through. The overall odds of a drug reaching approval starting from Phase I now sit near 7–8%. Efficacy issues not toxicity drive a large share of that attrition. Median capitalized R&D cost per approved drug, once you account for failures and cost of capital, now runs north of $700 million, and mean estimates are considerably higher still. Closing the Alignment Gap is one of the few competitive advantages left that doesn't require a bigger budget just better discipline.
The Development Alignment Gap
Successful development isn't really about generating data. It's about generating the right data, for the right audience, at the right time.
Most organizations start with something genuinely promising a strong hypothesis, a compelling mechanism, an innovative platform. The misalignment creeps in later, as that science moves toward regulatory review. An endpoint hits statistical significance without showing meaningful clinical benefit. A study population is broad enough to dilute a real treatment effect into noise. A biomarker that could have guided an early go/no-go decision shows up two years too late to matter. None of this requires anyone to make an obvious mistake. It happens through a series of individually defensible choices that drift, cumulatively, away from what regulators and payers are going to ask.
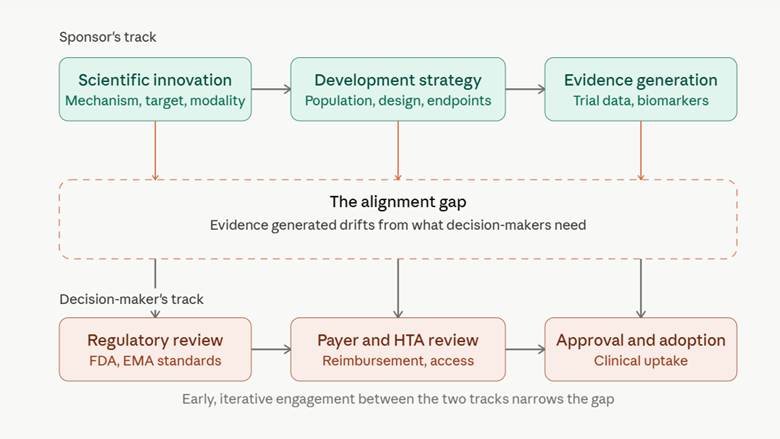
Sponsors move along their own track scientific innovation, development strategy, evidence generation while regulators, payers, and HTA bodies sit on a parallel track governed by review standards, reimbursement criteria, and adoption decisions. The gap opens wherever the first track gets ahead of, or simply diverges from, the second. Closing it isn't something you do once, near the end. It takes ongoing, iterative contact between the two tracks for the life of the program.
Case Studies: Two Trajectories
The two scenarios below are composites, built from patterns I've seen recur across multiple programs rather than any single client's story.
Scenario 1: The Program That Drifted
The first involved a mid-sized sponsor developing a treatment for a fibrotic disease. Their Phase IIb trial was well powered and hit its primary endpoint a continuous biomarker measuring tissue scarring with strong statistical significance. Walking into the End-of-Phase 2 meeting, the team was confident.
FDA's response wasn't what they expected: the agency had no established basis for believing that biomarker actually tracked with how patients felt or functioned day to day, and without that link, a clean p-value wasn't going to carry the program. The sponsor had generated rigorous evidence. It just wasn't evidence anyone downstream could act on. What followed was roughly 18 months retrofitting a patient-reported outcome measure into the confirmatory study a fix that would have cost a fraction of the time and money if someone had raised the question before the trial was designed around that endpoint in the first place.
Scenario 2: The Program That Aligned
The second sponsor, smaller and working in a rare neuromuscular indication, did something different. Before locking their pivotal trial design, they brought the proposed endpoint, population, and statistical plan to FDA in a Type B meeting and asked directly whether a functional outcome measure under development would be considered adequately validated in that population.
The agency's answer included something the sponsor hadn't fully considered on its own: a baseline severity threshold that would meaningfully sharpen the trial's ability to detect a real effect. The study that resulted was smaller and faster, and it landed on a pivotal readout the agency had effectively already signaled it would accept. Whether that single meeting alone explains the program's eventual approval is hard to say with certainty plenty else had to go right too but the team was unambiguous that it changed the trajectory of everything that came after.
The Problem is Often Not the Molecule
When a program fails, attention turns first to the product. Was the mechanism wrong? Was the target misunderstood? Was the therapy just not effective enough?
Those are fair questions, but they rarely tell the whole story. Plenty of programs spend years on protocol refinement and operational excellence only to discover, late, that stakeholders never agreed on what "meaningful clinical benefit" meant in the first place. Elsewhere, a real signal gets buried in a population that was too heterogeneous to find it, or a biomarker that could have guided enrollment arrives after the enrollment decisions are already made.
Call these what they are: failures of alignment, not failures of science.
Population Alignment
Genetics, imaging, molecular biology, and biomarker science have made one thing clear: most diseases are far more heterogeneous than we used to assume. Enroll too broad a population and a real treatment effect can get diluted past the point of detection not because the drug doesn't work, but because the trial wasn't built to find where it works.
Enrichment strategies help close that gap without engineering an artificially favorable cohort:
- Biomarker-guided enrollment
- Genetic stratification
- Imaging-based characterization
- Disease severity thresholds
- Prior treatment-response profiles
The Goal: The intent isn't to stack the deck toward a positive result. It's to enroll the exact population that can actually answer the clinical question the trial is asking.
Endpoint Alignment
Endpoint selection is one of the highest-consequence decisions in a development program, and it's easy to get subtly wrong. An endpoint can clear every statistical bar in the protocol and still fail to show meaningful clinical benefit. The reverse happens too, more quietly: a real benefit exists, but the endpoint chosen wasn't sensitive enough to catch it.
Strong programs anchor on one question before anything else gets decided: what evidence would convince a regulator, a clinician, and a patient that this therapy meaningfully improves outcomes? That question not precedent from a competitor, not statistical convenience should drive endpoint selection, balanced against clinical relevance, operational feasibility, and direct regulatory feedback.
Regulatory Alignment
Regulatory misalignment is one of the more preventable risks in development, which is exactly why it's so frustrating when it occurs. Sponsors who get this right build regulatory strategy into the program from day one rather than treating it as something to clean up once the science is settled. Every meaningful decision population, endpoints, statistical plan, biomarker strategy, safety monitoring, even manufacturing carries regulatory weight that compounds as the program advances.
The sponsors who do this well tend to show up to regulatory meetings with a specific kind of preparation:
- INTERACT or Pre-IND Meetings: Work best when the team arrives with a sharp, isolated question. Instead of asking for a generic program review, ask: "Does the agency agree this surrogate endpoint, used this way, in this population, could support approval?"
- Type B Meetings (Post-Phase II): Follow the same philosophy. The briefing package should isolate the one or two core assumptions the entire pivotal design rests on and ask the agency directly whether those assumptions hold up under scrutiny.
Treated this way, the meeting becomes a genuine pressure test before the trial design is locked in — not a bureaucratic formality on the way to a plan that was already decided.
Alignment Isn't Always Achievable All at Once
It would be convenient to say that enough foresight lets a sponsor satisfy every stakeholder simultaneously. That isn't always true, and a strategic framework does sponsors no favors pretending otherwise. FDA's evidentiary bar and a payer's or HTA body's bar for "meaningful benefit" overlap, but they are not identical.
A regulator might accept a well-validated surrogate that a payer won't reimburse against. A payer's preferred comparator might differ from the one that makes the most sense for a lean registration trial. In situations like these, alignment stops being about finding one design that pleases everyone and becomes about making an informed trade-off.
Sponsors must know ahead of time where regulatory and payer expectations will diverge, deliberately deciding which gap to leave open now and close later usually through a post-approval study or a separate health-economics analysis. This foresight doesn't make the tension disappear, but it ensures it is managed intentionally rather than discovered as a crisis after the pivotal trial reads out.
Evidence Generation as a Strategic Discipline
Rising costs and growing pressure for efficiency have turned evidence generation into a core strategic capability rather than an operational hand-off. The goal isn't just more data; it's data that is decision-relevant addressing efficacy, safety, patient selection, and market access as one connected ecosystem instead of four isolated workstreams.
Adaptive Development Strategies
Traditional designs lock in major assumptions before a single patient enrolls. Adaptive designs build in flexibility without sacrificing scientific or statistical integrity:
- Sample size re-estimation
- Population enrichment
- Adaptive randomization
- Seamless phase transitions
These tools don't make a wrong assumption less likely. What they do is lower the cost of being wrong by giving the program a structured, validated mechanism to course-correct mid-stream.
Biomarkers and Precision Evidence Generation
Biomarkers now shape patient selection, disease monitoring, and response prediction throughout a program's life cycle. Validated rigorously across analytical validity, clinical validity, clinical utility, and regulatory acceptance — they sharpen signal detection at every phase transition. They aren't a shortcut around careful trial design; rather, when deployed early, they make that design fundamentally more efficient.
Expedited Pathways Reward Early Planning, Not Late Opportunism
Fast Track, Breakthrough Therapy, Accelerated Approval, and RMAT offer real efficiency gains for serious conditions. However, the FDA does not treat them as procedural shortcuts to apply for late in the game. The agency views them as evidence-planning opportunities that should shape endpoint strategy, biomarker validation, and confirmatory study design from the start. Sponsors who treat a designation as a badge to collect, rather than a framework to build the program around, frequently find themselves negotiating confirmatory trial requirements under far more pressure than necessary.
The Role of Artificial Intelligence
AI is increasingly built into protocol development, patient matching, safety surveillance, data analysis, and medical writing. In well-run programs, it is measurably improving both execution speed and data operational quality.
Still, it is worth being precise about what actually changes: AI sharpens clinical judgment, regulatory expertise, and strategic thinking it does not substitute for them. The core challenge of closing the Alignment Gap generating evidence that answers the critical questions a regulator or payer will ask is a strategic and scientific problem before it is a computational one. AI can help a well-aligned team move faster, but it cannot make a poorly aligned strategy sound.
Looking Ahead
The next decade will bring deeper precision medicine, expanded digital health tools, more real-world evidence, and sharper predictive analytics. All of that will continue to accelerate how evidence gets generated.
What won't change is the foundational question sitting underneath it all: does the totality of evidence support a favorable benefit-risk profile for the intended population? No amount of technical sophistication answers that question for you. It still must be asked correctly, early, and often.
Key Takeaways for Sponsors
- Begin with the end in mind: Align your evidence strategy with downstream stakeholder needs starting on Day 1 — not after the pivotal data is already locked.
- Prioritize structural alignment: Focus early on population and endpoint alignment to give your molecule's true clinical signal its best chance of being seen.
- Engage transparently and iteratively: Treat regulatory bodies as pressure-tests for your core design assumptions, using targeted questions rather than passive document reviews.
- Lower the cost of assumptions: Deploy adaptive designs and robustly validated biomarkers to mitigate strategic risk, not to defer hard decisions.
- Elevate evidence strategy: Place evidence generation directly on the executive leadership agenda, rather than siloing it exclusively within clinical operations.
- Anticipate divergent expectations: Explicitly map the gaps between regulatory approval and payer reimbursement early and plan the specific mechanisms to bridge them.
Conclusion
The organizations that consistently succeed aren't necessarily the ones with the largest budgets or the broadest pipelines. They're the ones that hold scientific objectives, clinical evidence, regulatory expectations, payer requirements, and patient needs in deliberate alignment for the entire life of the program.
Innovation will always matter, but it is rarely enough on its own. The therapies that successfully reach patients are those built on excellent science coupled with an evidence strategy designed, from day one, with approval, adoption, and real-world utility already in view.
About Salience Clinical LLC
If your organization is looking to identify or bridge its own development gaps, an independent strategic review is often the fastest way to surface misalignment before it impacts your timelines. Salience Clinical, LLC works with sponsors on clinical development strategy, FDA regulatory planning, biomarker integration, and approval-focused program design.
Contact us to discuss your pipeline.

About the Author
Denis Katz, MD is a physician executive with nearly two decades of leadership experience across clinical development, medical affairs, regulatory strategy, and evidence generation in pharma, biologics, regenerative medicine, diagnostics, and neuroscience. Through Salience Clinical LLC, he advises emerging and established organizations on closing the Development Alignment Gap to bring promising therapies to patients more efficiently.